For some time now, proteins known as amyloids have been implicated in the onset and advance of Alzheimer’s and other diseases, such as type 2 diabetes. One of the curious aspects of linking these proteins to diseases is that it seems to be primarily unusual protein folding and assembly that leads to disease. This is especially so in the case of Alzheimer’s and cerebral amyloid angiopathy, where mutations such as the Iowa mutant are associated with familial inheritance and early onset of the disease. Patients carrying the mutation develop neuritic plaques and large deposits of the mutant protein in cerebral blood vessels. Exactly how the protein does so much damage has been the subject of intense recent research, including these findings by researchers from The University of Chicago, the National Institutes of Health, and the Illinois Institute of Technology. With the help of the BioCAT 18-ID beamline at the APS, the team used x-ray diffraction, electron microscopy, and nuclear magnetic resonance (NMR) spectroscopy to show how the mutant and normal proteins differ with respect to folding and assembly. What the research group found goes a long way toward explaining how the mutant protein is able to create so much havoc in tissues and how -amyloids relate to disease.
The researchers focused on the β-amyloid coded by the Iowa mutation, which differs from the normal protein by only one amino acid. They found that the mutant protein, known as D23N-Aβ40, forms fibrils much more rapidly than the normal protein and does so without first undergoing the lag phase observed for the normal protein. X-ray diffraction patterns from the mutant protein showed cross-β patterns and other attributes that differed from the normal protein. Observing the mutant protein using electron microscopy, the team found that it occurs in more than one morphotype (organisms that are classified together on the basis of similar physical features without knowledge of their genetic relationship).
Further investigations using NMR spectroscopy showed that only a minority of the mutant protein fibrils looked like the typical β-amyloid protein fibrils, which exhibit a parallel β-sheet structure. Thus, a majority of the mutant protein fibrils looked quite different from the normal fibrils. Further examination of the mutant fibrils indicated that they are put together differently and had a smaller average diameter when compared to the normal fibrils.
In perhaps the most exciting of the new findings presented by the investigators, a majority of the mutant fibrils exhibited a structure consistent with antiparallel β-sheets and are thus assembled quite differently from the parallel β-sheet of the normal protein. This clear difference in orientation between the mutant and normal molecules represents one of the first truly distinguishing characteristics of the mutant protein and will certainly lead to better ways of detecting and treating the resulting disease states.
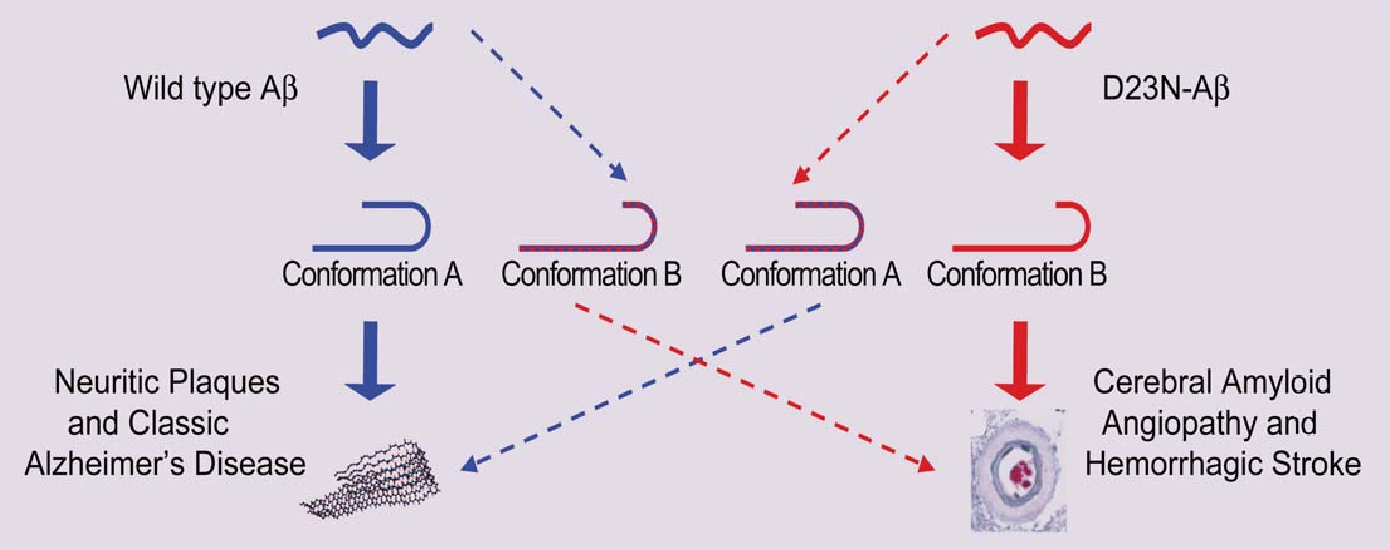
Using these new data, the research team proposed a link between aberrant assembly and tissue pathology of the patients carrying the mutation for D23NAβ40 (Fig. 1). Further work is planned to answer questions about how exactly the mutant protein assembles to produce some molecules that look like the normal protein, even though most of them do not. Previous work by the research group showed that examining the mechanisms for bends or folds between sheets holds promise for explaining these differences and allowed them to construct a model consistent with their current results. Additional questions include how the substitution of a single amino acid in the mutant protein allows it to form the very different anti-parallel β-sheet structure. And the large protein deposits characteristic of the disease in patients carrying the Iowa mutation, as well as the frequency with which β-amyloid protein is transported into the blood vessel wall, may also be related to how and when the fibrils are assembled.
The work reported by the research team points to two characteristics of proteins that can never be ignored: the amino acid sequence and how the protein is assembled. In the case of the β-amyloid proteins, changing one amino acid creates a mutant protein that can assemble quite differently and lead to diseases such as Alzheimer’s, an understanding of which may lead to the development of new therapeutic options.
See: Robert Tycko, Kimberly L. Sciarretta, Joseph P.R.O. Orgel, and Stephen C. Meredith, “Evidence for Novel β-Sheet Structures in Iowa Mutant β-Amyloid Fibrils,” Biochemistry-US 48, 6072 (2009). DOI: 10.1021/bi9002666
This work benefited from the support of the NSF Research Collaborative Network, “Fibernet” (MCB-0234001). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Based on an APS press release by Mona Mort.