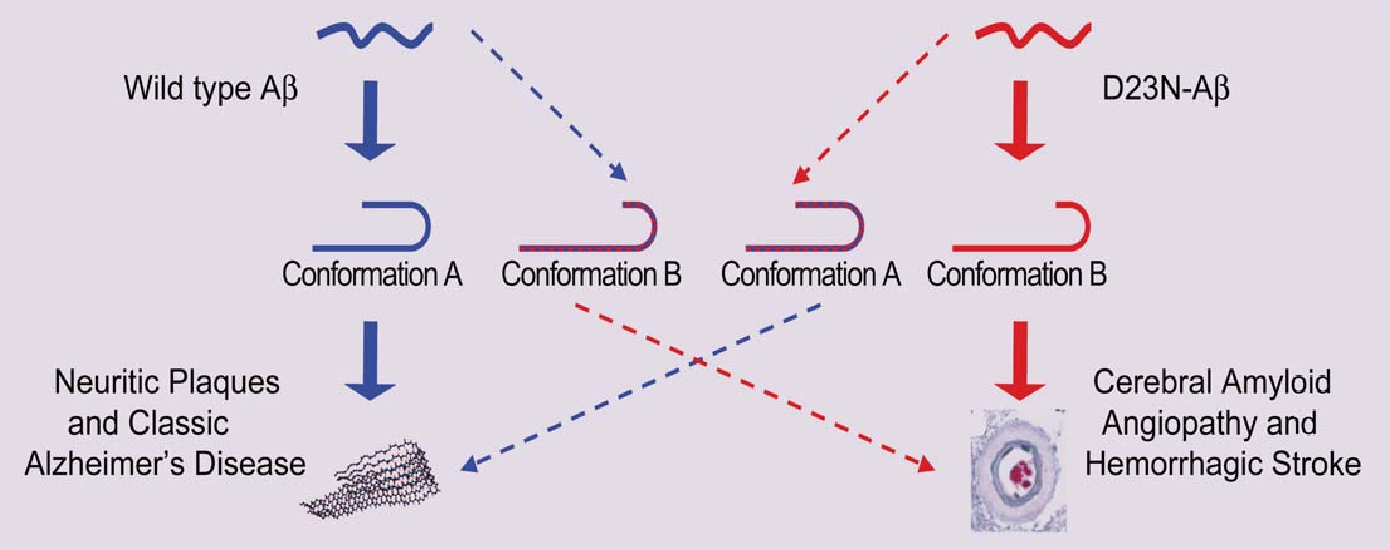
Increased Brain Iron Coincides with Early Plaque Formation in a Mouse Model of Alzheimer’s Disease

Early and correct diagnosis of Alzheimer’s disease (AD) is important for reasons that go beyond correct diagnosis and treatment of symptoms. These reasons include more time to make critical life decisions, planning for future care, and maximizing the safety of the person with Alzheimer’s disease and their family. New scientific results relevant to the diagnosis and treatment of AD have been obtained by researchers utilizing the U. S. Department of Energy’s Advanced Photon Source (APS) at Argonne National Laboratory and National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory, and published in the journal NeuroImage. This work points to the use of elevated brain iron content, already observed in late-stage AD, as a potential tool for early diagnosis. Since the disease is usually diagnosed only in later stages after cognitive symptoms appear and treatment may not be effective, a method for early detection would be a …