A Super-relaxed Myosin State to Offset Hypertrophic Cardiomyopathy
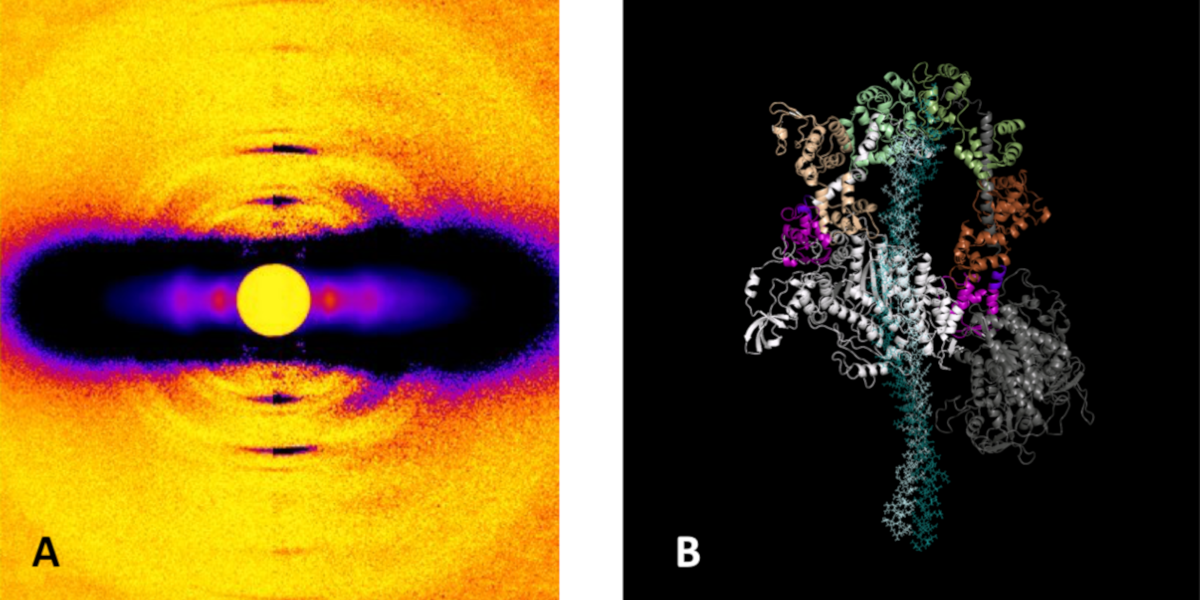
At its most basic level, the proper functioning of the heart depends upon the intricate interaction of proteins that trigger, maintain, and control the muscular contractions and relaxations of this vital organ. Disruption of those interactions can cause serious pathologies such as hypertrophic cardiomyopathy (HCM). Such disruptions can originate with mutations in the primary motor protein involved in heart contraction, ß-cardiac myosin, which can alter the rate of ATP hydrolysis and have been hypothesized to destabilize its super-relaxed state (SRX). Researchers investigated the stabilizing action of mavacamten, a cardiac drug currently in phase 3 clinical trials, on the ß-cardiac myosin super-relaxed state and its possible therapeutic effects on HCM. Their work, which included electron microscopy and low-angle x-ray diffraction imaging at the U.S. Department of Energy’s Advanced Photon Source …